 中文
中文 
產品細節圖:
| 產品編號 | bs-10589R |
| 英文名稱 | Collagen II |
| 中文名稱 | Ⅱ型膠原蛋白抗體 |
| 別 名 | Collagen II alpha 1; COL2A1; COL2A1 protein; collagen, type II, alpha 1; collagen alpha-1(II); type II collagen; alpha-1 type II collagen; alpha1 type II collagen; Col2a1; AOM; Cartilage collagen; Chondrocalcin; COL11A3; Collagen alpha 1(II) chain precursor; Collagen II alpha 1 polypeptide; Collagen type II alpha 1 (primary osteoarthritis spondyloepiphyseal dysplasia congenital); MGC131516; SEDC; CO2A1_HUMAN. |

|
Specific References (7) | bs-10589R has been referenced in 7 publications.
[IF=4.263] Wu X et al.The role of Ca 2+ in acid-sensing ion channel 1a-mediated chondrocyte pyroptosis in rat adjuvant arthritis.(2018) Lab Invest. WB ; Rat .
[IF=3.76] Shi, Yang, et al. "Hypoxia combined with spheroid culture improves cartilage specific function in chondrocytes." Integrative Biology 7.3 (2015): 289-297. Rat.
[IF=2.784] Ma C et al.Isolation and biological characteristic evaluation of a novel type of cartilage stem/progenitor cell derived from Small?tailed Han sheep embryos.Int J Mol Med. 2018 Jul;42(1):525-533. ICF ; Sheep.
[IF=2.348] Ren et al. Treatment of rabbit intervertebral disc degeneration with co-transfection by adeno-associated virus-mediated SOX9 and osteogenic protein-1 double genes in vivo. (2013) Int.J.Mol.Me. 32:1063-8 WB ; Rabbit.
[IF=0] Xu et al. Treatment with SiMiaoFang, an anti-arthritis chinese herbal formula, inhibits cartilage matrix degradation in osteoarthritis rat model. (2013) Rejuvenation.Re. 16:364-76 IHC-P ; Rat.
[IF=0] Yang et al. Isolation and biological characterization of tendon-derived stem cells from fetal bovine. (2016) In.Vitro.Cell.Dev.Biol.Ani. 52:846-56 IF(ICC) ; Rat.
[IF=] Luo P et al. IL-37b alleviates inflammation in the temporomandibular joint cartilage via IL-1R8 pathway. Cell Prolif. 2019 Sep 27:e12692. IHC-P ; Rat.
|
| 研究領域 | 腫瘤 細胞生物 免疫學 |
| 抗體來源 | Rabbit |
| 克隆類型 | Polyclonal |
| 交叉反應 | Human, Mouse, Chicken, Dog, Pig, Cow, Horse, Rabbit, |
| 產品應用 |
WB=1:500-2000 ELISA=1:500-1000 IHC-P=1:100-500 IHC-F=1:100-500 ICC=1:100-500 IF=1:100-500 (石蠟切片需做抗原修復) not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. |
| 分 子 量 | 117kDa |
| 細胞定位 | 細胞外基質 分泌型蛋白 |
| 性 狀 | Liquid |
| 濃 度 | 1mg/ml |
| 免 疫 原 | KLH conjugated synthetic peptide derived from human Collagen II:231-330/1487 |
| 亞 型 | IgG |
| 純化方法 | affinity purified by Protein A |
| 儲 存 液 | 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. |
| 保存條件 | Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. |
| PubMed | PubMed |
| 產品介紹 |
Collagens are highly conserved throughout evolution and are characterized by an uninterrupted "Glycine-X-Y" triplet repeat that is a necessary part of the triple helical structure. For these reasons it is often extremely difficult to generate antibodies with specificities to collagens. The development of type specific antibodies is dependent on NON DENATURED three dimensional epitopes. This may result in diminished reactivity of some antibodies with denatured collagen or formalin fixed, paraffin embedded tissues. Type II collagen is a fibrillar collagen found in cartilage and the vitreous humor of the eye. Collagen type II is essential for the normal embryonic development of the skeleton, for linear growth and for the ability of cartilage to resist compressive forces. Mutations in this gene are associated with achondrogenesis, chondrodysplasia, early onset familial osteoarthritis, SED congenita, Langer Saldino achondrogenesis, Kniest dysplasia, Stickler syndrome type I, and spondyloepimetaphyseal dysplasia Strudwick type. In addition, defects in processing chondrocalcin, a calcium binding protein that is the C propeptide of this collagen molecule, are also associated with chondrodysplasia. There are two transcripts identified for this gene. Function: Type II collagen is specific for cartilaginous tissues. It is essential for the normal embryonic development of the skeleton, for linear growth and for the ability of cartilage to resist compressive forces. Subunit: Homotrimers of alpha 1(II) chains. Subcellular Location: Secreted, extracellular space, extracellular matrix. Tissue Specificity: Isoform 2 is highly expressed in juvenile chondrocyte and low in fetal chondrocyte. Post-translational modifications: Probably 3-hydroxylated on prolines by LEPREL1 (By similarity). Proline residues at the third position of the tripeptide repeating unit (G-X-P) are hydroxylated in some or all of the chains. Proline residues at the second position of the tripeptide repeating unit (G-P-X) are hydroxylated in some of the chains. The N-telopeptide is covalently linked to the helical COL2 region of alpha 1(IX), alpha 2(IX) and alpha 3(IX) chain. The C-telopeptide is covalently linked to an another site in the helical region of alpha 3(IX) COL2. DISEASE: Defects in COL2A1 are the cause of spondyloepiphyseal dysplasia congenital type (SEDC) [MIM:183900]. This disorder is characterized by disproportionate short stature and pleiotropic involvement of the skeletal and ocular systems. Defects in COL2A1 are the cause of spondyloepimetaphyseal dysplasia Strudwick type (SEMD-STR) [MIM:184250]. A bone disease characterized by disproportionate short stature from birth, with a very short trunk and shortened limbs, and skeletal abnormalities including lordosis, scoliosis, flattened vertebrae, pectus carinatum, coxa vara, clubfoot, and abnormal epiphyses or metaphyses. A distinctive radiographic feature is irregular sclerotic changes, described as dappled in the metaphyses of the long bones. Defects in COL2A1 are the cause of achondrogenesis type 2 (ACG2) [MIM:200610]; also known as achondrogenesis-hypochondrogenesis type II. ACG2 is a disease characterized by the absence of ossification in the vertebral column, sacrum and pubic bones. Defects in COL2A1 are the cause of Legg-Calve-Perthes disease (LCPD) [MIM:150600]; also known as Legg-Perthes disease or Perthes disease. LCPD is characterized by loss of circulation to the femoral head, resulting in avascular necrosis in a growing child. Clinical pictures of the disease vary, depending on the phase of disease progression through ischemia, revascularization, fracture and collapse, and repair and remodeling of the bone. Defects in COL2A1 are the cause of Kniest dysplasia (KD) [MIM:156550]; also known as Kniest syndrome or metatropic dwarfism type II. KD is a moderately severe chondrodysplasia phenotype that results from mutations in the COL2A1 gene. Characteristics of the disorder include a short trunk and extremities, mid-face hypoplasia, cleft palate, myopia, retinal detachment, and hearing loss. Defects in COL2A1 are a cause of primary avascular necrosis of femoral head (ANFH) [MIM:608805]; also known as ischemic necrosis of the femoral head or osteonecrosis of the femoral head. ANFH causes disability that often requires surgical intervention. Most cases are sporadic, but families in which there is an autosomal dominant inheritance of the disease have been identified. It has been estimated that 300,000 to 600,000 people in the United States have ANFH. Approximately 15,000 new cases of this common and disabling disorder are reported annually. The age at the onset is earlier than that for osteoarthritis. The diagnosis is typically made when patients are between the ages of 30 and 60 years. The clinical manifestations, such as pain on exertion, a limping gait, and a discrepancy in leg length, cause considerable disability. Moreover, nearly 10 percent of the 500,000 total-hip arthroplasties performed each year in the United States involve patients with ANFH. As a result, this disease creates a substantial socioeconomic cost as well as a burden for patients and their families. Defects in COL2A1 are the cause of osteoarthritis with mild chondrodysplasia (OACD) [MIM:604864]. Osteoarthritis is a common disease that produces joint pain and stiffness together with radiologic evidence of progressive degeneration of joint cartilage. Some forms of osteoarthritis are secondary to events such as trauma, infections, metabolic disorders, or congenital or heritable conditions that deform the epiphyses or related structures. In most patients, however, there is no readily identifiable cause of osteoarthritis. Inheritance in a Mendelian dominant manner has been demonstrated in some families with primary generalized osteoarthritis. Reports demonstrate coinheritance of primary generalized osteoarthritis with specific alleles of the gene COL2A1, the precursor of the major protein of cartilage. Defects in COL2A1 are the cause of platyspondylic lethal skeletal dysplasia Torrance type (PLSD-T) [MIM:151210]. Platyspondylic lethal skeletal dysplasias (PLSDs) are a heterogeneous group of chondrodysplasias characterized by severe platyspondyly and limb shortening. PLSD-T is characterized by varying platyspondyly, short ribs with anterior cupping, hypoplasia of the lower ilia with broad ischial and pubic bones, and shortening of the tubular bones with splayed and cupped metaphyses. Histology of the growth plate typically shows focal hypercellularity with slightly enlarged chondrocytes in the resting cartilage and relatively well-preserved columnar formation and ossification at the chondro-osseous junction. PLSD-T is generally a perinatally lethal disease, but a few long-term survivors have been reported. Defects in COL2A1 are the cause of multiple epiphyseal dysplasia with myopia and conductive deafness (EDMMD) [MIM:132450]. Multiple epiphyseal dysplasia is a generalized skeletal dysplasia associated with significant morbidity. Joint pain, joint deformity, waddling gait, and short stature are the main clinical signs and symptoms. EDMMD is an autosomal dominant disorder characterized by epiphyseal dysplasia associated with progressive myopia, retinal thinning, crenated cataracts, conductive deafness. Defects in COL2A1 are the cause of spondyloperipheral dysplasia (SPD) [MIM:271700]. SPD patients manifest short stature, midface hypoplasia, sensorineural hearing loss, spondyloepiphyseal dysplasia, platyspondyly and brachydactyly. Similarity: Belongs to the fibrillar collagen family. Contains 1 fibrillar collagen NC1 domain. Contains 1 VWFC domain. SWISS: P02458 Gene ID: 1280 Database links: Entrez Gene: 1280 Human Entrez Gene: 12824 Mouse Omim: 120140 Human SwissProt: P02458 Human SwissProt: P28481 Mouse Unigene: 408182 Human Unigene: 2423 Mouse Unigene: 10124 Rat Important Note: This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
| 產品圖片 |
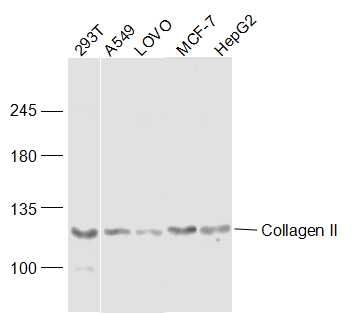
Sample:
293T(Human) Cell Lysate at 30 ug A549 (Human) Cell Lysate at 30 ug LOVO (Human) Cell Lysate at 40 ug MCF-7 (Human) Cell Lysate at 30 ug HepG2 (Human) Cell Lysate at 30 ug Primary: Anti-Collagen II (bs-10589R) at 1/300 dilution Secondary: IRDye800CW Goat Anti-Rabbit IgG at 1/20000 dilution Predicted band size: 117 kD Observed band size: 117 kD 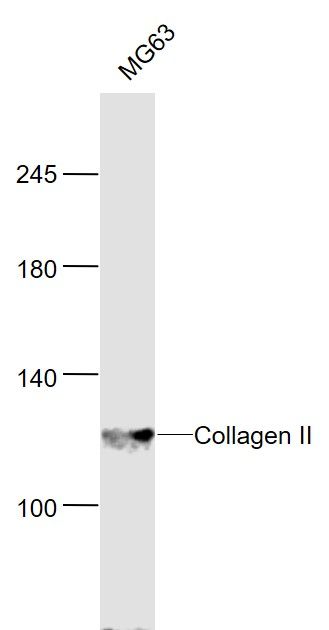
Sample:
MG63 (Human) Cell Lysate at 30 ug Primary: Anti-Collagen II (bs-10589R) at 1/300 dilution Secondary: IRDye800CW Goat Anti-Rabbit IgG at 1/20000 dilution Predicted band size: 117 kD Observed band size: 117 kD 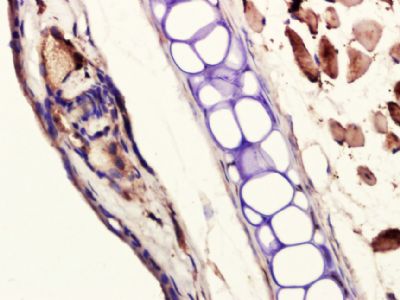
Paraformaldehyde-fixed, paraffin embedded (mouse ear); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (Collagen II) Polyclonal Antibody, Unconjugated (bs-10589R) at 1:400 overnight at 4°C, followed by a conjugated secondary (sp-0023) for 20 minutes and DAB staining.

Tissue/cell: Mouse embryo tissue; 4% Paraformaldehyde-fixed and paraffin-embedded;
Antigen retrieval: citrate buffer ( 0.01M, pH 6.0 ), Boiling bathing for 15min; Block endogenous peroxidase by 3% Hydrogen peroxide for 30min; Blocking buffer (normal goat serum,C-0005) at 37∩ for 20 min; Incubation: Anti-Collagen II Polyclonal Antibody, Unconjugated(bs-10589R) 1:100, overnight at 4∑C, followed by conjugation to the secondary antibody(SP-0023) and DAB(C-0010) staining |
 暫時沒有說明書
暫時沒有說明書
1.《Multifunction Sr, Co and F co-doped microporous coating on titanium of antibacterial, angiogenic and osteogenic activities》
作者:Jianhong Zhou ,Lingzhou Zhao 影響因子:4.259
期刊:《Scientific Reports 6, Article number: 29069 (2016)》 PMID:27353337
作者:Jianhong Zhou ,Lingzhou Zhao 影響因子:4.259
期刊:《Scientific Reports 6, Article number: 29069 (2016)》 PMID:27353337
| 用戶名: | (可為空) |
| E-mail: | *(必填) |
| 評價等級: |





|
| 評論內容: |
*(必填)
|





